Boys With Rett Give Us Clues About MeCP2’s Function
 Rett Syndrome is caused by a variety of mutations in the MeCP2 protein, but in some instances, MeCP2’s end is missing. A graduate student in Developmental Biology at the Baylor College of Medicine in Houston, Steven Baker, who is also in the medical scientist training program, was sifting through the clinical literature on boys with such mutations when he noticed that a tiny difference in how much of the protein’s tail is shortened—by just three amino acids—seemed to make the difference between decades of life (albeit with Rett-like deficits) and death in infancy.
Rett Syndrome is caused by a variety of mutations in the MeCP2 protein, but in some instances, MeCP2’s end is missing. A graduate student in Developmental Biology at the Baylor College of Medicine in Houston, Steven Baker, who is also in the medical scientist training program, was sifting through the clinical literature on boys with such mutations when he noticed that a tiny difference in how much of the protein’s tail is shortened—by just three amino acids—seemed to make the difference between decades of life (albeit with Rett-like deficits) and death in infancy.
Baker asked his adviser, Huda Zoghbi, whether she thought those extra few amino acids could so dramatically change the clinical progression of Rett.

"I don’t know," Zoghbi, Howard Hughes Medical Institute Investigator and director of the Jan and Dan Duncan Neurological Research Institute at Texas Children’s Hospital, recalls telling him, because other genetic factors could be contributing the dramatic difference in the progression of Rett. “The only way to know is to make mice that have the two different mutations.”
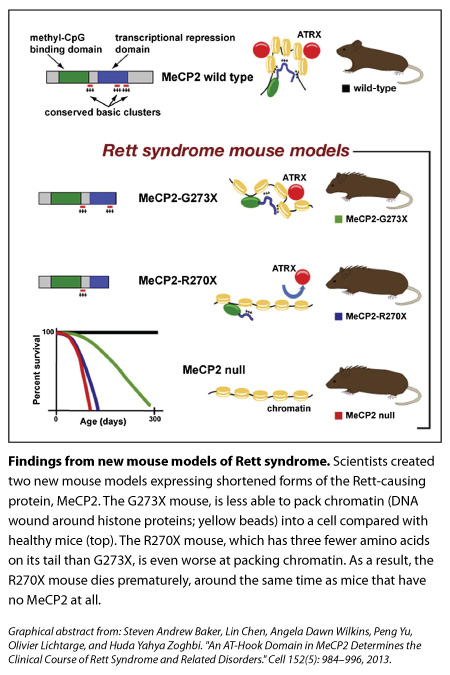
So they did. One mouse had the end of its MeCP2 cut off at the 270 mark (‘R270X’ mice) while another’s protein was shortened at the 273 mark (‘G273X’ mice). The stories of these mice were reminiscent of the boys Baker had noticed. The R270X mice died prematurely, around the same time as mice with no MeCP2. In contrast, the G273X mice, with their extra three amino acids, survived longer and showed symptoms later, although these features became more severe and the mice died before healthy mice did.
What do those amino acids do? In trying to find out, the scientists have refined their understanding of how MeCP2 works. Their results are published this week in Cell.
A first look at DNA binding:
Researchers know that MeCP2 binds throughout the genome, coating the DNA in neurons more heavily than in other cells, but what exactly it does after that is less clear. It’s thought to turn genes on or off, or alter the overall structure of DNA.
Zoghbi’s team thought that the two truncated forms of MeCP2 might bind to DNA differently — in a way that would help explain the different clinical progressions of the boys — but when they initially looked at several spots within the genome they saw that both forms bound to those spots similarly.
In fact, looking more broadly across the genome the group found that overall binding of the MeCP2 was pretty much the same, and it looked normal. (The latter wasn’t too surprising, though, because the front end of MeCP2 was already known to bind to DNA.) Both mutations also interfered with the normal ability of MeCP2 to repress genes.
Looking more closely at gene expression at various time points in brain development, however, the group found a key difference in the two mutants: at 4 weeks of age, a small group of genes was improperly expressed in the R270X mutant but not in the G273X mutant. Interestingly, most of those genes eventually became misregulated in the G273X group by 9 weeks.
The hook from the animal kingdom:
Meanwhile, the scientists asked evolutionary biologist, Olivier Lichtarge, a Professor at Baylor College of Medicine who uses computational tools to study the evolution of protein sequences, to help them compare the sequences of the protein’s tail across different species (the idea being that any regions of the protein that were highly similar across species might be important for the protein’s function). “We worked with Angela Wilkins in the Lichtarge lab and asked, ‘Is there something in this domain that’s really unique?" says Zoghbi, who is also a Professor at Baylor College of Medicine.
They found that three clusters of MeCP2’s tail were highly conserved across fish, frog, rat, mouse, and human. Shortening MeCP2 at the 273 mark removed the third of those conserved clusters, whereas cutting the tail at the 270 mark deleted the second and third clusters.
What are these clusters in MeCP2’s tail and what do they do? It turns out that they’re called AT-hooks. In 2005, a study by Adrian Bird’s group described the first of those three AT-hooks (which is the one still present in both of Zoghbi’s new models), though its function was unclear.
AT-hooks are regions of a protein that are already well known to bind DNA, however, so the team went back to the idea that the two truncated proteins might differ from one another in how they attached to the genome, even though their initial results had shown that binding was similar. Using a different assay, they found that missing the second AT-hook domain impaired the ability of the R270X to steadily interact with certain sequences in the genome.
The interaction between MeCP2 and DNA:
Our genomes are wound tightly around spool-like proteins called histones; the DNA and histones (together, called chromatin, which looks like beads on a string) are then packed in even more so that it can all fit inside cells. Using an experimental model of compaction in vitro, the team found results suggesting that R270X mice (which, remember, are missing an two AT-hooks instead of one) don’t pack up chromatin as well as 273X mice do.
The initial finding that both mutated forms of MeCP2 bind to DNA are still important though, Zoghbi says. “It tells us that the major binding to DNA happens. That’s the first step.” The researchers think that once the front end of MeCP2 sits on DNA, the AT-hook clusters on its tail come in manipulate the DNA further, likely bending or altering the structure to help it pack further into the cell.
“There were hints previously that MeCP2 might cause a change in the overall conformation of DNA. The new study is probably the most direct evidence,” says Howard Hughes Medical Institute Investigator Gail Mandel, at the Oregon Health and Science University in Portland, who was not involved with the study.
The protein partner ATRX:
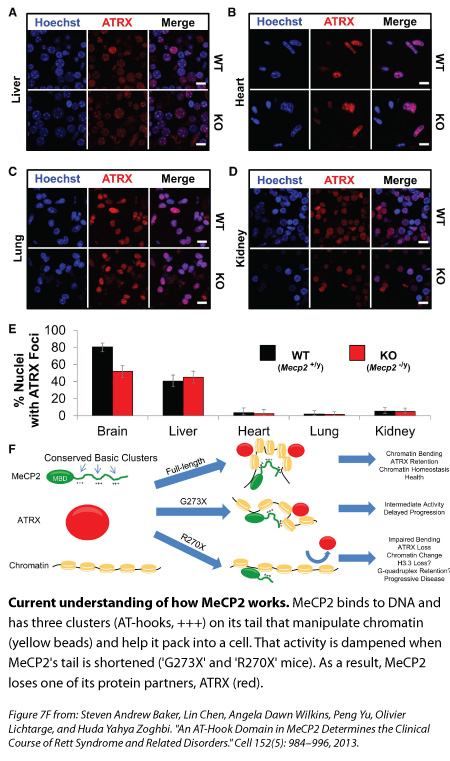
When the MeCP2 sits on DNA, and likely alters the way it packs into a cell, there are other molecular partners that come and join it. One of those is the protein ATRX—whose mutations have been linked to Alpha-thalassemia mental retardation syndrome—and Adrian Bird’s lab has previously shown that its interaction with DNA is disrupted in mice missing MeCP2. Zoghbi’s team decided to look at this protein in their new mutant mice.
Compared with the healthy mice and the G273X mice, ATRX goes missing from the tightly packed DNA of neurons earlier in life for the R270X mouse, and this loss mirrors the quicker onset of Rett symptoms. “To us, that was really interesting,” Zoghbi says, “because this change is not because the neurons are sick, it’s because you don’t have MeCP2 functioning properly.”
Studying female mutant mice that are missing a copy of MeCP2, the researchers found those brain cells with no MeCP2 also had less ATRX bound to tightly packed DNA compared to controls. In female mutants with too much MeCP2, an excess of ATRX latched on to DNA. MeCP2’s absence from liver and non-brain organs didn’t affect ATRX binding in those organs, suggesting that MeCP2 has a mechanism that’s specific for the brain.
Future directions:
Taken together, these results suggest that in the brain, the AT-hook clusters on MeCP2’s tail are manipulating DNA in a way that’s crucial for the other protein partners to bind and do their jobs, Zoghbi says.
“This new paper is beginning to shed light on the complexity of this interaction between MeCP2 and ATRX,” says Mandel. In addition, “we don’t know all the other proteins that bind to MeCP2, but the guess would be that there are likely more partners affecting whether genes are on or off.”
Zoghbi’s team hopes to understand how shortening MeCP2’s tail changed chromatin structure without dramatically changing gene expression — as well as the mutation’s affect on brain activity. They also plan to do biochemical and molecular experiments to figure out where ATRX is going and what it’s doing when its distribution is altered in the brain cells of the MeCP2 mutants.
For Zoghbi, the new findings underscore the importance of going back to patients to look for clues about MeCP2’s function. In 1999, Zoghbi first showed that various mutations in MeCP2 caused Rett. “Here we are 14 years later, some of these human mutations are teaching us lessons,” she says. “The variety you get and the breadth of human features you can dissect and go back and study in the mouse are really very humbling.”
