Rare diseases don’t cure themselves. Developing genetic medicines is a complex, expensive effort with many moving parts. We are the organization orchestrating this global effort, and here is our plan to cure Rett syndrome.
The Goals of Roadmap to Cures 2.0

Why is Roadmap 2.0 Essential?



“The promise of Gene Therapy is that it addresses the fundamental root cause of a disease. But it’s still in early stages. There’s so much innovation left on the table. You can’t rely on yesteryears technology, you have to constantly be innovating.”
Where Rett Cures Begin
Our children’s futures depend on a rich and diverse pipeline of genetic medicine programs. Keeping this pipeline flourishing and producing results requires deep scientific and industry experience, laser focus on the mission, and an appropriate level of funding.

Where We've Been
In 2007, Prof. Adrian Bird shocked the scientific and Rett community by showing that Rett symptoms in mice models disappeared upon restoration of MECP2. Within months Monica launched RSRT, with Prof. Bird at her side, with the sole purpose of accelerating cures. Explore our journey so far.

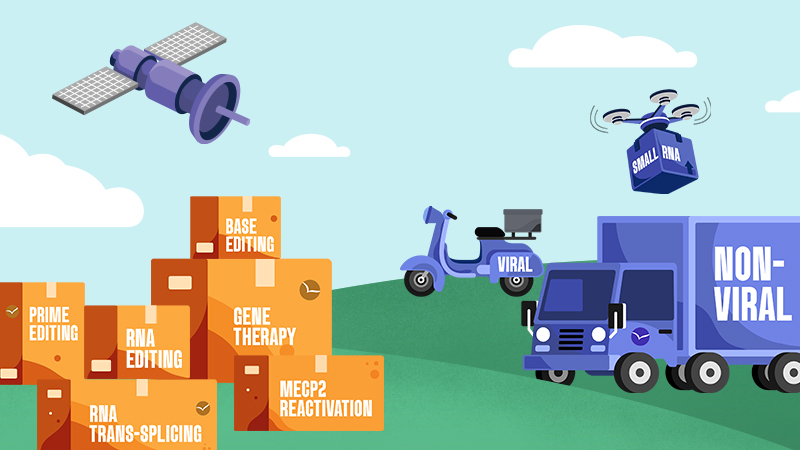
Genetic medicines are made up of two parts: the cargo
which corrects for the mutation, and the
delivery technology that gets the cargo into cells.
The Approaches
Cargo

The introduction of healthy genes into the body to compensate for mutated genes. This approach is the most advanced of the genetic medicines and is already in clinical trials for Rett. This approach is relevant for all types of MECP2 mutations.
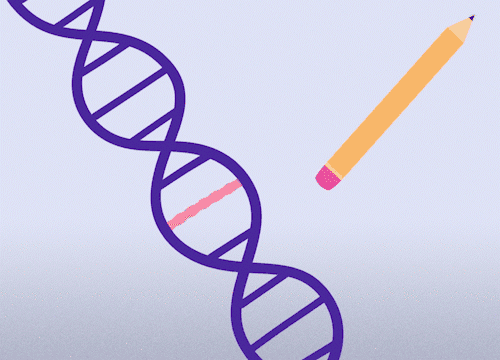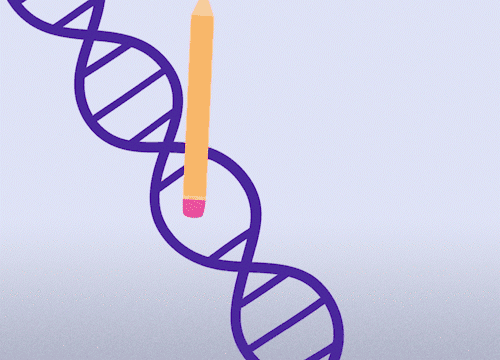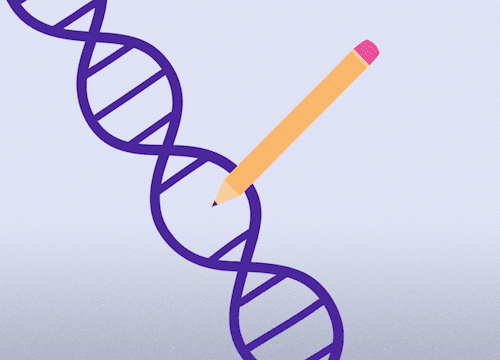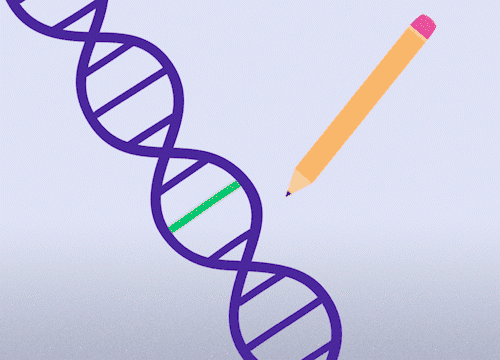
This approach enables precise changes to the DNA sequence at specific locations and can be used to correct point mutations (typos in a single nucleotide base).

This approach extends beyond base editing and addresses a wide array of mutations including insertions and deletions. It can correct multiple mutations that are located near each other on the gene. In time, prime editing will address all MECP2 mutations.
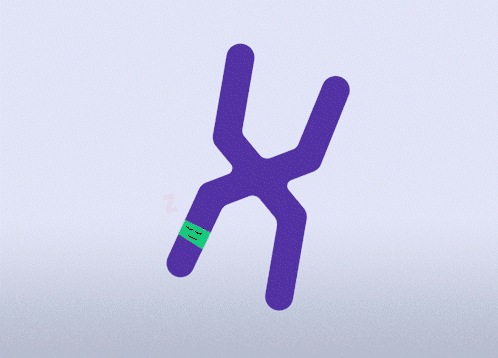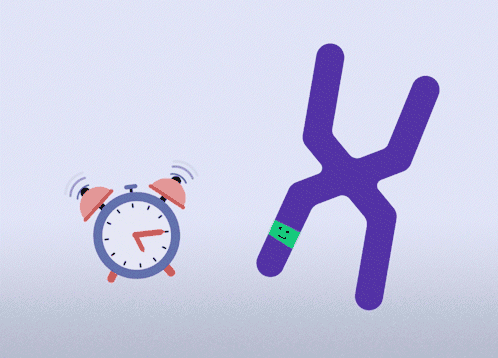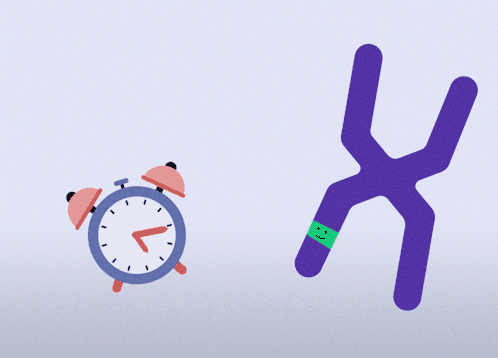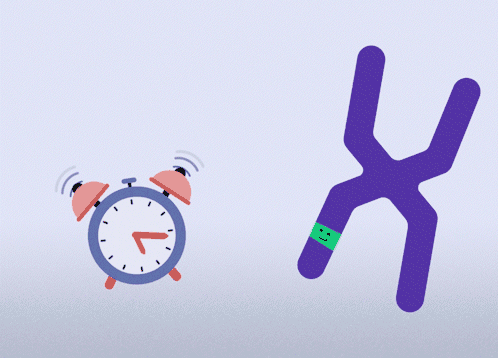
All females have two X chromosomes, one active and one inactive. In cells where the mutated gene is active, there is a healthy backup gene on the inactive X chromosome. The goal of this approach is to awaken the healthy, silenced gene.This approach is relevant for all types of MECP2 mutations.
This approach converts one nucleotide to another within the RNA sequence and can be used for specific point mutations.

MECP2 consists of 4 exons. This approach replaces mutated exons with healthy exons. This approach is relevant for all MECP2 mutations.
Delivery

Viral
Viruses are very efficient at delivering their own genetic material into cells during infection. This function can be harnessed to allow viruses to serve as delivery vehicles for genetic cargos.

Non-Viral
Delivery vehicles extend beyond viruses to include approaches such as lipid nanoparticles and exosomes. Non-viral delivery offers a number of important advantages.

Small RNA
Small RNA are able to self deliver and do not need viral or non viral delivery. They have been used clinically for many years with much success and can therefore advance rapidly into the clinic.
Tracking
Collecting and analyzing comprehensive and accurate data on individuals with Rett improves their clinical care and ensures that clinical trials deliver clear and meaningful results. This, ultimately, drives the development of transformative genetic medicines which is why it's so important that we have mechanisms in place to gather this information.

“The one thing we've learned about how to build a cure for genetic disease is we have to put as many different technologies for those diseases into the clinic and learn as much as we can.”
Genetic Medicines Pipeline by 2028
Our efforts have catalyzed a rich pipeline of MECP2 targeted genetic medicines.
This pipeline is dynamic and will be updated as new data emerges.

Into the Clinic



What About My Child?
Some genetic medicine approaches, like gene therapy, apply to all mutations while others
are mutation specific. Find out which approach is relevant for your loved one.

The Business of Genetic Medicines
The spark for a new medicine typically happens with a discovery in an academic laboratory. Testing in cell cultures is followed with testing in animal models. With encouraging data in hand, intellectual property (IP) is filed. Scientists sometimes start a company that licenses the IP from the academic institution. More often, however, the institution markets the IP to biopharma companies. Once the IP is licensed, the biopharma company begins work on a disease indication.
The divide between academia and biopharma is often referred to as the “Valley of Death'' because most discoveries never make the transition. With a deep understanding of the genetic medicine space and strong networks we eliminate the “ Valley of Death” by bridging the divide between them.
We award funding strategically to elite academic scientists to ensure a robust pipeline of cargos and delivery vehicle options. As these work streams mature, we pair the best combinations of collaborators, while simultaneously lining up biopharma partners to advance the programs into the clinic.
Developing genetic medicines is a risky business both on the science side and business side. The best way to mitigate that risk is by taking more shots on goal.
The development of genetic medicines for Rett does not happen on its own. It’s an effort that must be driven, brokered and monitored closely. It requires deep industry and business development experience, tenacity and skin in the game.
News & Updates






Rett Genetic Medicines Summit 2025
Over the course of three days in September we convened leading scientists, clinicians, and regulators to explore every corner of the genetic medicines landscape. The leadership from over 45 biopharma companies had a unique opportunity to hear about the state of Rett genetic medicine programs, with the ultimate goal of inspiring and motivating them to add the disorder to their pipelines.
This year’s gathering underscored RSRT’s role as both convener and catalyst, bringing brilliant minds together while never losing sight of the children and families at the heart of our community.

