The Story Behind Mini-MeCP2

In May 2022 Taysha Gene Therapies made the exciting announcement that they will start a phase 1/2 clinical trial of gene therapy for Rett syndrome. Their plan to restore MeCP2 to the nervous system by introducing the functional protein into cells that lacked it, was a welcome development in the struggle to find a Rett cure. It was closely followed by an announcement from Neurogene that they too intended to trial gene therapy for Rett syndrome. Gene “replacement” of this kind involves hijacking a virus to deliver a functional MECP2 gene into the brain. A distinctive feature of the Taysha trial is that, rather than using the entire MeCP2 coding sequence, they have chosen a cut-down version which is much shorter. This article explains how the “mini-MeCP2” came about. (Note: I have no involvement with Taysha or the proposed trial).
When my research group came up with the mini-MeCP2 idea, we were not primarily thinking of it as a therapeutic vector, but more as a way of finding out how the MeCP2 protein actually works. All human proteins have evolved to perform functions that help make a healthy individual. MeCP2 is no exception, but working out exactly what it does has not been easy. Tantalising clues came from looking at the types of mutation that cause the condition. As background, proteins consist of chains of amino acids of which there are 20 different kinds. For the protein to work properly, the right amino acids have to be in precisely the right order. Mutations mess this up, either in a surgically discrete way, by substituting one amino acid in the chain for the wrong one, or drastically — by causing loss of chunks of the chain. Whether the mutations are discrete or drastic, the end result is the same: a serious shortage of working MeCP2 — which leads to Rett syndrome.
No protein acts alone, so one way of getting at function is to look for partners that work with it. In the case of MeCP2 we had known for a long time that one of its partners is DNA — specifically DNA that has been methylated. In 2013, lab member Matt Lyst found a second important partner: a huge multi-protein complex called NCoR. Using biochemistry, we narrowed down both these interaction domains to discrete regions of the MeCP2 protein chain which we called the MBD (Methyl-CpG Binding Domain) and the NID (NCoR Interaction Domain) respectively. Crucially, the vast majority of Rett mutations inactivate one or both of these domains. Particularly informative were the subtle Rett-causing mutations which pinpoint amino acids in the chain that can’t be changed at all. Our findings suggested a simple hypothesis to explain what MeCP2 does: it serves as a bridge between sites of DNA methylation in the genome and the NCoR corepressor. In other words, it recruits NCoR to the genomic DNA of brain cells — particularly neurons where MeCP2 is extremely abundant. Mutations which disrupt either end of the MeCP2 bridge prevent recruitment from happening, leading to Rett syndrome (see cartoon below).
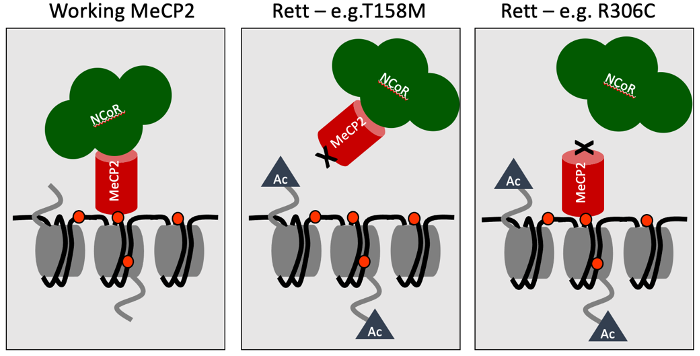
Scientific hypotheses at their best are disposable scientific tools that provoke experiments with the potential to prove you wrong. For example, the bridge hypothesis predicts that all Rett syndrome mutations should impact the MBD or the NID, but in fact some Rett mutations are not close to the MBD or the NID, suggesting that something else might be going on. When Jacky Guy, a member of my lab, looked into these mutations in more detail, however, it turned out that they caused the mutated MeCP2 protein to be unstable and consequently degraded. Thus, even though these mutations did not directly affect the MBD or the NID, they did lead to almost complete loss of MeCP2. The bridge hypothesis survived this and other tests, but we still sought a “killer” experiment with the potential to annihilate what had become our favoured explanation for MeCP2 function.
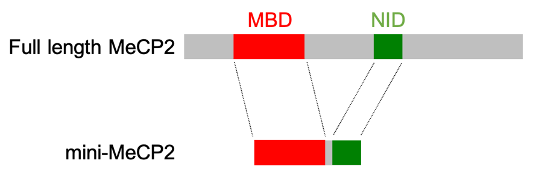
The idea was simple — replace the natural MECP2 gene in mice with a truncated version that retained the MBD and the NID but lacked the remaining two-thirds of the protein. If we were right, these two domains alone should be able to do the job of full length MeCP2. The reason why it seemed a risky experiment is that the sequence of amino acids in MeCP2 protein is very highly conserved during evolution. That is, the chain has remained virtually the same in all mammals and pretty much the same even in fish and frogs. As mutations are occurring all the time, one might expect that over the many millions of years of evolution that separate, for example, mice and humans the sequence of the amino chain would drift, as it does for many other proteins. The fact that this has not happened to MeCP2 suggests that that the whole thing has to be “just so”, or it doesn’t work properly. What hope was there that chopping off most of such a protein (see diagram below) would be compatible with normal life?
The idea hung around for quite a while waiting for someone willing to take on a high-risk project. Fortunately, PhD student Becky Tillotson picked up the gauntlet and showed that mice whose MECP2 gene had been replaced by one encoding mini-MeCP2 were indeed remarkably healthy. Without any MeCP2, mice die a couple of months after birth, but mini-MeCP2 mice had a lifespan of more than a year and resembled mice with no mutation. Jim Selfridge, a long-time member of my lab, suggested that we test whether switching on the mini-gene after mice had become sick could rescue their problems. This experiment was based on the “classic” reversal experiments done by Jacky Guy showing that delayed restoration of MeCP2 could reverse the severe neurological problems in mice suffering from Rett syndrome. As it turned out, Becky found that mini-MeCP2 could do the same thing, offering striking support for the view that the MBD and the NID alone can accomplish most of what MeCP2 does in the brain. As the icing on the cake, we collaborated with Stuart Cobb’s lab, which was still in Glasgow at that time (he has since joined Edinburgh University), to see whether delivering the gene for mini-MeCP2 in an AAV9 virus could also rescue Rett mice. Once again, there were impressive improvements, suggesting that this version of the protein might be therapeutically useful.
Why use a cut-down version of MeCP2 for gene therapy rather than the whole thing? Our original study noted two potential advantages. Firstly, the payload capacity of AAV9 vectors is very limited, so making the gene shorter creates more space for adding other bits of DNA to optimise or regulate the amount of protein product. Secondly, we gained the impression that well-established toxicity caused by too much MeCP2 is less likely with mini-MeCP2. Subsequent unpublished work in Stuart Cobb’s laboratory has shown that in fact too much mini-MeCP2 is also toxic in mice, so, as with full-length MeCP2, an excessive therapeutic dose needs to be avoided. Both planned trials take steps to make sure MeCP2 amounts are kept in check. Despite inevitable uncertainties, there is good reason to hope that mini-MeCP2 can provide clinical benefit — provided it can be delivered to enough nerve cells. The latter proviso represents a big unknown: is AAV9 capable of brain-wide delivery in humans? The consensus view is that we will almost certainly need a better viral vector before long, and many academic and industrial labs are working hard to find one. Looking on the bright side, though, even a small amount of MeCP2 gives a significant improvement in Rett mice. Here’s hoping the Taysha and/or Neurogene initiatives can give us the encouragement that we crave.
