New Insight Into MECP2 Mutation Hotspot

Rett syndrome-causing missense mutations cluster in two regions of MECP2. One of these regions - the methyl-CpG binding domain (MBD) - has been extensively characterized and helps MeCP2 bind DNA. The other region is called the NCoR/SMRT interaction domain (NID) and recruits a gene-silencing complex, but its function has remained comparatively mysterious.

Why are mutations in the NID so debilitating?
Dr. Adrian Bird and colleagues at the University of Edinburgh explored the function of the NID and identified a molecular partnership that is crucial for brain function. Their work is published this week in the journal Proceedings of the National Academy of Sciences.
They Report Three Key Findings
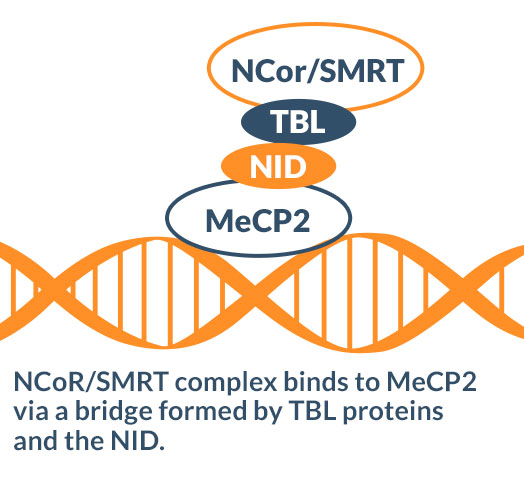
- MeCP2 binds to a gene-silencing complex at the NID through proteins called TBLs.
- Missense mutations in the NID (like R306C, K304E, P302R, and K305R) disrupt TBL binding by destabilizing or interfering with the structure of the TBL-MeCP2 complex. For example R306C, which accounts for 5% of Rett syndrome cases, prevents the formation of an essential salt bridge and destabilizes the 3D conformation of the complex.
- Individuals with TBL mutations that disrupt binding to MeCP2 are reported to display features of Rett syndrome, including intellectual disability, developmental delay, and ASD.
Conclusions and Future Directions
This study shows that the interaction between MeCP2, TBLs, and DNA is crucial for brain function - disruption of this complex causes severe disability. But why? Dr. Matthew Lyst, an author on the paper, acknowledges, “this is a huge unanswered question for the field.” He says their next steps are to investigate which parts of the complex mediate MeCP2 function and to develop treatments that stabilize the interaction between mutant proteins.
