Learning from Patients
R ett Syndrome doesn’t usually run in the family. Researchers led by Alessandra Renieri at the University of Siena in Italy encountered two exceptional cases: one pair of sisters with the same mutation in the Rett-causing gene MECP2, and a second pair with identical deletions within the gene.
ett Syndrome doesn’t usually run in the family. Researchers led by Alessandra Renieri at the University of Siena in Italy encountered two exceptional cases: one pair of sisters with the same mutation in the Rett-causing gene MECP2, and a second pair with identical deletions within the gene.
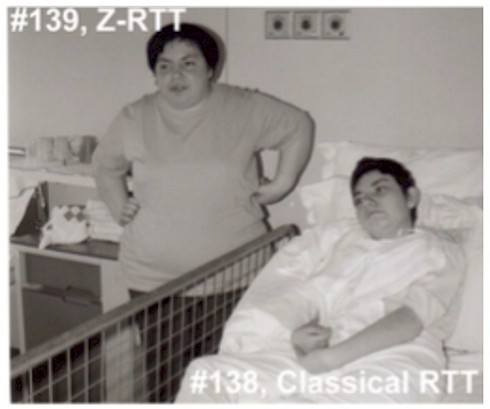
Despite having the same mutations in MECP2, the sisters represented the clinical spectrum of the disorder. For each pair, one sister had classical Rett Syndrome—she was unable to speak or walk or use her hands—while the other had a milder form of the disorder (called Zapella) and could talk using short phrases, walk and retained some hand function. Researchers describe these four women and possible genetic reasons why the severities of their symptoms were so different, in a PLOS ONE paper published a few months ago.
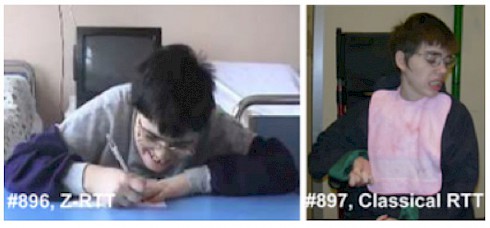 It’s not surprising that girls with Rett Syndrome generally show a wide range of symptoms. That’s partly because a mutated copy of the MECP2 gene is located on only one of two X chromosomes in a female cell; the other copy is healthy. One X chromosome becomes inactive in each cell early in development. In rare cases when many cells express the healthy copy of MECP2, women show a milder form of Rett. In the new study, however, both pairs of women were similar in how many of their maternally or paternally derived X chromosomes were inactivated, suggesting that something else might explain the differing severities of their disease.
It’s not surprising that girls with Rett Syndrome generally show a wide range of symptoms. That’s partly because a mutated copy of the MECP2 gene is located on only one of two X chromosomes in a female cell; the other copy is healthy. One X chromosome becomes inactive in each cell early in development. In rare cases when many cells express the healthy copy of MECP2, women show a milder form of Rett. In the new study, however, both pairs of women were similar in how many of their maternally or paternally derived X chromosomes were inactivated, suggesting that something else might explain the differing severities of their disease.
Hypothesizing that other genes could contribute to these differences, the scientists sequenced a small proportion of the women’s genomes (about 1%) that is thought to code for proteins. (This strategy, called exome sequencing, is a less costly and less burdensome in terms of data analysis compared with whole-genome sequencing, and in recent years it has been shown to identify previously unknown genes for rare, inherited disorders, such as Freeman-Sheldon syndrome.)
 The team located 112 genetic variants on 108 genes that were exclusive to the women with classical Rett. A subset of these variations, about 10 to 20, is believed to be relevant to impaired protein function in Rett, based on what’s already known about them.
The team located 112 genetic variants on 108 genes that were exclusive to the women with classical Rett. A subset of these variations, about 10 to 20, is believed to be relevant to impaired protein function in Rett, based on what’s already known about them.
These genes are involved in a range of functions. Interestingly, both women with classic Rett have variants on at least six genes that have been previously linked to oxidative stress. (The two people with Zappella had variants on three.) In a follow-up study, Renieri’s group found that the women with classical Rett, but not the two with Zappella, showed molecular signs of oxidative stress compared with healthy controls. But the link between MECP2 mutations and oxidative stress is still unknown, the authors note.
The women with Zappella had exclusive variants in 80 genes, but none of these were shared by both. Some genes are linked to immune function, and the variants may be involved in protection from a more severe phenotype, says Renieri, a professor of medical genetics.
Although exome sequencing will continue to bear genetic clues on the variability of Rett Syndrome, the meaning of these variants will need further study, Renieri says. “I’m not sure now that all the variants we describe in the paper are relevant,” she admits. “In the next few years we will learn better how to interpret these results.”
Renieri’s group hopes to sequence the exomes of more people with severe and mild Rett Syndrome, to understand their genetic similarities and differences. It is easier to compare the genes of sisters because their genomes are 50% identical. But because sisters with Rett are so rare, they will need to compare unrelated patients, she says.
